Vaginitis is one of the most common health-related issues that affects women worldwide. It can be caused by bacterial, fungal or protozoal infections. Vulvovaginal candidiasis (VVC) is the second most common cause of vaginitis, accounting for one-third of all cases in women of reproductive age.1 In the USA, VVC results in approximately 1.4 million outpatient visits annually.2
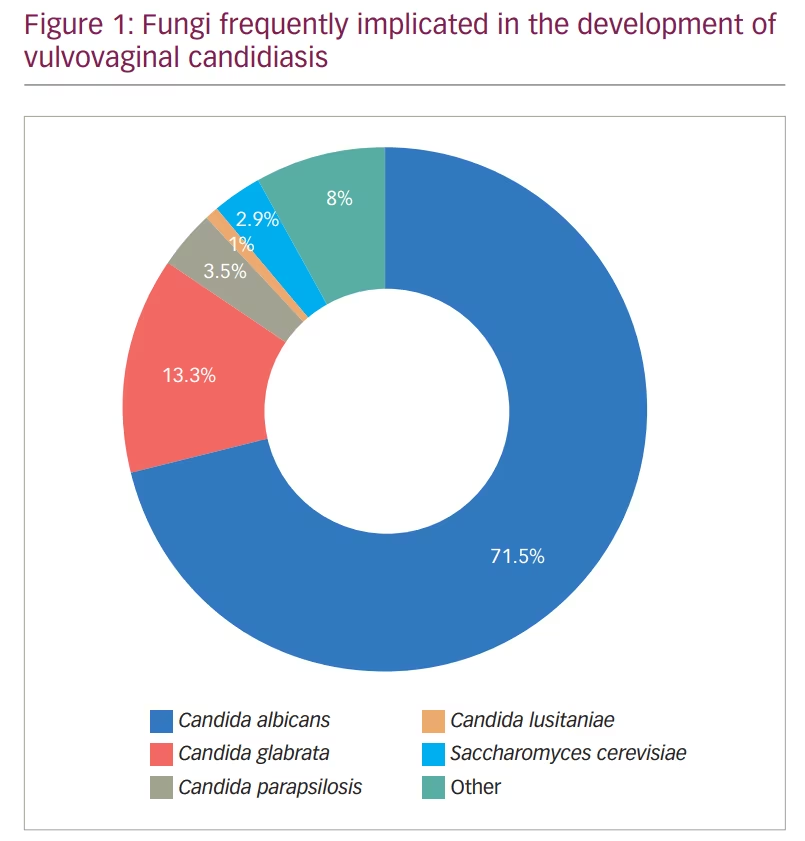
Among the causative fungi, Candida albicans was shown to be the main culprit in causing this infection;3,4 however, infections with other non-albicans Candida – such as C. glabrata and C. krusei – have also been observed (Figure 1).3,4 The interaction between Candida and the vaginal mucosa leads to a local inflammatory response, which may present clinically with several symptoms such as vaginal pruritus, soreness, dyspareunia, external dysuria and vaginal discharge. On the other hand, vaginal colonization by Candida may also be asymptomatic.5–7
VVC is well recognized as a multifactorial disease (Table 1). The causative organism, Candida, can directly invade the tissue via its hyphal filaments or secretion of extracellular virulence effectors including aspartyl proteases, phospholipases and candidalysin.8,9 Biofilm formation is another virulence factor attributed to Candida; however, its role in the pathogenesis of VVC is still unclear. In this regard, some in vitro and in vivo vaginitis models suggest that biofilm formation protects Candida from antifungals and the host immune system response, resulting in sustained local inflammation. Establishing a confirmed role for biofilm formation in VVC in patients remains to be demonstrated.10–12 Disruption of the vaginal microbiota, especially reduction of the Lactobacillus species, which inhibits Candida adherence to the vaginal epithelium (90–95%), may influence VVC pathogenesis.13–16 Host-related factors also influence VVC pathogenesis including: use of contraceptives, post-menopause hormone replacement therapy, pregnancy, uncontrolled diabetes mellitus, altered immune status, use of steroids, and antibiotic overuse.17–21 All of these factors may increase the risk of VVC development by various mechanisms and affect propensity for recurrence.17–22

The CDC has defined recurrent vulvovaginal candidiasis (RVVC) as three or more episodes of symptomatic VVC in less than 1 year, which is estimated to occur in around 5% of women.23 According to the study by Denning et al., the estimated annual number of RVVC cases worldwide is approximately 138 million, and is projected to increase to almost 158 million by 2030.24
Several factors increase the likelihood for RVVC. For example, mannose-binding lectin 2 gene mutation (an important element in immune response against microbes) has been reported frequently in patients with RVVC compared with healthy women,25–28 supporting a role for genetic factors in the development of RVVC. Furthermore, immunosuppression has been reported to be associated with higher incidence of RVVC.29,30 Although antifungal resistance, including resistance of C. albicans and C. auris, is becoming a public health concern worldwide,31,32 resistance of fungi responsible for VVC is not common. However, it is prudent to monitor the antifungal incidence of resistance and institute antifungal stewardship in this disease to avoid future development of resistance. In this review, we discuss past commonly used VVC treatments, recently approved antifungals and promising future alternative therapies.
Treatment options
Currently, two treatment approaches are available for VVC: topical and systemic therapy. Both treatment modalities have been considered equally effective in the treatment of uncomplicated VVC, which can be defined as: 1. infrequent episodes (≤3 episodes/year); 2. mild-to-moderate signs/symptoms; 3. probable infection with C. albicans; 4. healthy, non-pregnant individuals; and 5. immunocompetent individuals.33,34 Although some studies have suggested that topical treatment offers several advantages and unique features that may favour this therapeutic approach, oral regimens are often preferred by physicians and patients.35 The appeal of oral therapy has been attributed to this therapeutic modality being a single dose, which – unlike topical products – is simple and efficient.
Either topical or systemic treatment of VVC has been predominantly limited to the azole class of antifungals.36 Although azole antifungals are effective and well tolerated by most patients, several factors may limit their utility in some patients, such as intolerance to the topical formulations, side effect profiles and drug–drug interactions.37 Additionally, systemic fluconazole has been reported to be associated with an increased incidence of spontaneous abortion, especially when administrated in high-dose formulations (i.e. >150 mg/dL).38–41 Furthermore, there have been reports of an association between first trimester oral fluconazole and foetal malformation.38,42,43 Thus, in general, the use of oral fluconazole during pregnancy is discouraged. In patients for whom oral fluconazole is not an option, topical azoles and other agents such as nystatin or boric acid become the only available alternative drugs.44
Introduction of new antifungals to manage VVC, with different mechanisms of action compared with the currently available treatment options, may potentially help overcome the limitations associated with these treatments. This calls for the need to develop new treatments that effectively treat this infection and reduce the recurrence rate, while producing fewer side effects.
Vulvovaginal candidiasis treatment over the past decade
Systemic treatment
Oral fluconazole is the most commonly used antifungal to treat VVC. It acts by inhibiting ergosterol biosynthesis, a major component of fungal cell membranes, targeting the cytochrome P450-dependent enzyme lanosterol demethylase. This, in turn, causes depletion of ergosterol and accumulation of 14α-methylated sterols in the fungal cell membrane, leading to disruption of both the structure of the membrane and several of its functions, such as nutrient transport and chitin synthesis. This culminates in inhibition of fungal growth and proliferation, and eventual cell lysis and death.45
Generally, fluconazole is effective and well tolerated by most patients; however, because of its cross-interaction with the human cytochrome P450, side effects – including vomiting, diarrhoea, rash and increased liver enzymes – may occur when combined with some drugs (due to drug–drug interactions). Another side effect, although rare, is QTc prolongation, which can occur with oral fluconazole therapy in some patients.46–51 The main advantage of fluconazole is that it is administrated as a 150 mg single oral dose, which is more convenient for patients.23 This is true when managing a case with acute VVC; however, an extended maintenance therapy may be required in chronic cases to reduce the recurrence incidence.44 In this regard, the current treatment regimen recommended by the Centers for Disease Control and Prevention (CDC) for the management of RVVC includes induction therapy in the form of topical azole applications for 7–14 days or 100–200 mg of oral fluconazole every 72 hours for a total of three doses, which should be followed by 6 months of maintenance therapy of oral fluconazole (weekly dose of 100–200 mg).23 Although this regimen was shown to be effective, it has some limitations. For example, a recurrence rate of over 50% has been reported after stopping the weekly fluconazole, treatment can fail against resistant species, and long-term fluconazole carries an increased risk of side effects, such as xerosis, alopecia and fatigue.52
Another alternative to fluconazole is the use of itraconazole, recommended for patients with acute VVC, dosed as 100 mg orally 2 × 2 capsules daily for 1 day or 1 × 2 capsules daily for 3 days. Additionally, ketoconazole (200 mg twice daily for 5 days) is another alternative azole that has demonstrated good efficacy in the management of VVC.44,53
These observations clearly indicate that new treatments with fewer side effects may improve patients’ compliance to the regimen and, consequently, enhance the cure rate and reduce the frequency of recurrence.
Topical treatment
Several factors should be considered when choosing an antifungal agent for topical treatment of VVC, including the ability of the drug to penetrate deeply into the vaginal epithelium where invasive Candida hyphae reside, resulting in a local antifungal effect.54 Additionally, as the normal vaginal pH is acidic (ranges between 3.8 and 5.0), it is important that topical antifungal should be effective at low pH. In this regard, a reduction in the antifungal susceptibility of fungi isolated from VVC patients was observed at a low pH level in in vitro studies,55–57 resulting in an increased minimum inhibitory concentration (MIC) observed among resistant strains when tested against azole antifungals at a low pH level. Thus, the impact of low pH on the activity of antifungals should be considered when selecting or developing a new agent.55–57
Azole antifungals are also used in topical formulations (e.g. cream, ointment and suppositories) to treat VVC. The CDC recommends several over-the-counter medications for the treatment of VVC (Table 2). This recommendation includes clotrimazole 1–2% or miconazole 2–4% creams that can be used daily for 3–14 days. Additionally, vaginal suppositories are an alternative option that falls into the same category and includes miconazole 100, 200 and 1,200 mg vaginal suppositories dosed daily for 7, 3 or 1 day, respectively. Finally, tioconazole 6.5% ointment is also available for VVC treatment in a single application therapy. Other azole agents such as butoconazole and terconazole may also be used as therapeutic options; however, they require prescription by a physician.23,58 One of the main advantages of topical versus systemic azoles is the limited toxicity associated with topical use.
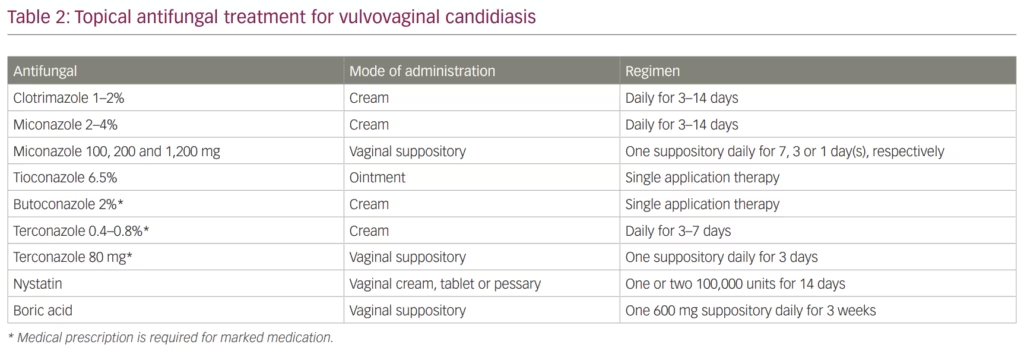
Beyond azoles, two alternative topical antifungals that can be used to treat VVC are topical nystatin and boric acid. Nystatin belongs to the polyene antifungal class that binds to ergosterol resulting in formation of pores in the fungal cell membrane causing cytoplasmic leakage, acidification, cell lysis and eventual death of the fungus.45 In animal models, nystatin exhibited an immunomodulatory effect that provided protection against C. albicans infection.59 Nystatin is usually administered in the form of vaginal tablets at 100,000 or 200,000 units for 6 or 14 days.44 Although the use of nystatin has decreased since the introduction of azole antifungal agents, it has some advantages over the azoles, especially when treating vaginitis caused by C. glabrata.60 The main side effects of nystatin that may limit its use are rash, itching and burning sensation.
In a clinical trial that included 293 patients with RVVC, treatment with either nystatin suppository (n=152) or oral fluconazole (n=141) resulted in mycological cure rates following the initial treatment course of 78.3% and 73.% for nystatin- and fluconazole-treated groups, respectively.60 At the end of 6 months of maintenance therapy, 80.7% of the nystatin group demonstrated mycological cure compared with 70.2% in the fluconazole group. Although both antifungals had similar cure rates against C. albicans (84.0% versus 81.8% for nystatin and fluconazole, respectively), nystatin had higher cure rate against C. glabrata compared with fluconazole (64.3% versus 12.5%, respectively). Similar results were also observed in another clinical trial, which included 46 patients with VVC treated with nystatin or fluconazole (23 patients in each treatment group). The antifungals demonstrated a cure rate of 74.0% and 87.0% for nystatin vaginal cream and oral fluconazole, respectively.61 However, unlike the first study, fluconazole had better activity against C. glabrata compared with nystatin. Thus, both antifungals demonstrate efficacy in the treatment of VVC. Consequently, nystatin can be considered as an alternative option to fluconazole in cases where oral fluconazole is contraindicated, or failure of other topical azole therapy occurs.
Boric acid is a compound of boron, oxygen and hydrogen, with the formula B(OH)3, that is used topically to treat VVC. The mechanism of action of boric acid is not completely understood; however, it is reported to inhibit both hyphal transformation (a known virulence factor) and biofilm formation of C. albicans, and arrests fungal growth.62 Boric acid has the advantage of being effective against azole-resistant non-albicans Candida species with fewer local side effects.63 Thus, it is recommended to be used in the management of RVVC in cases that fail to respond to azole therapy.23 However, because of its embryotoxic effect and the negative effect on fertility, the European Chemicals Agency has issued a warning against the application of boric acid, restricting its use to non-pregnant women in exceptional cases.64
Recently approved antifungals for the treatment of vulvovaginal candidiasis
Ibrexafungerp
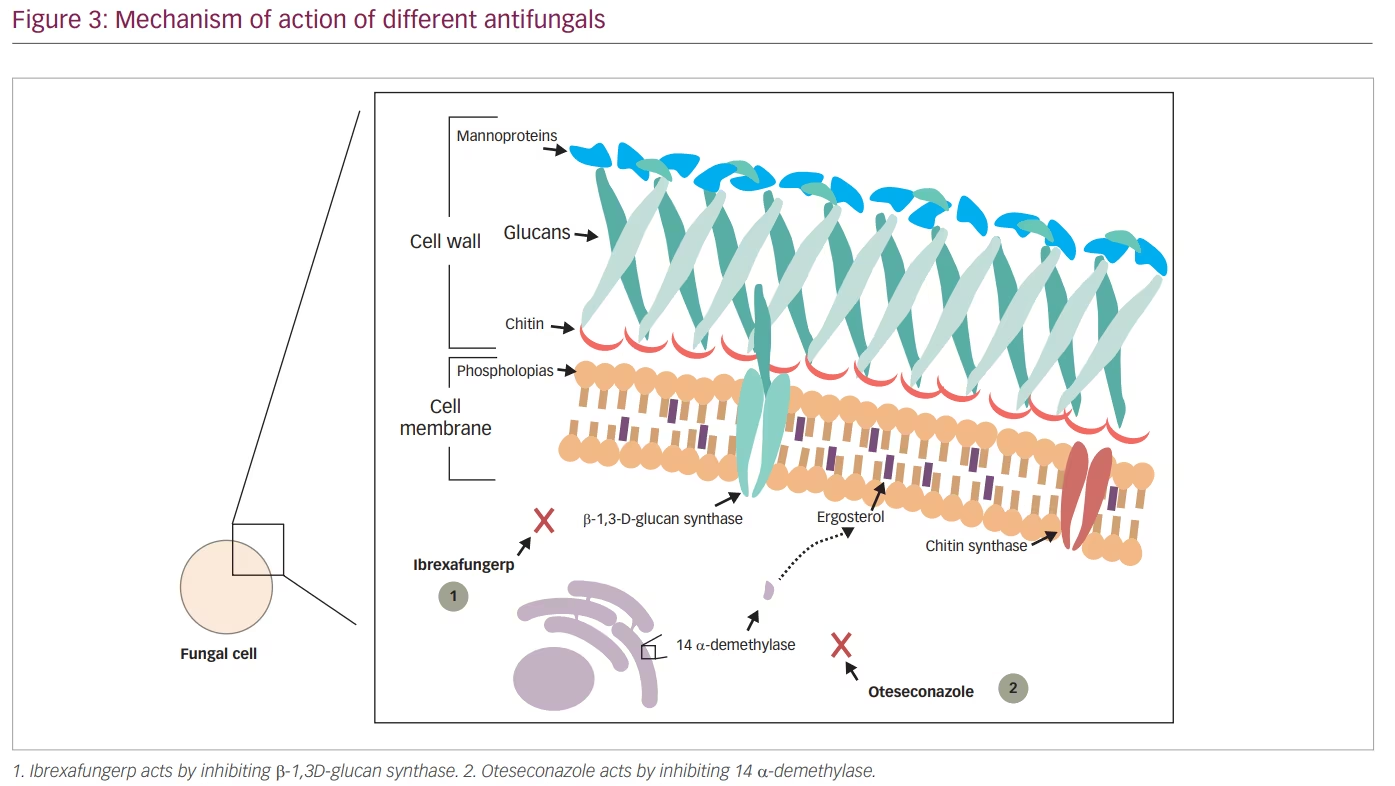
Echinocandins were shown to be effective in eliminating infections caused by Candida spp. that were resistant to azole antifungals. However, because echinocandins can only be administrated by intravenous infusion, their use in infections such as VVC was limited. Ibrexafungerp (IBX, previously SCY-078), a triterpenoid class antifungal, solves this issue by acting using a similar mechanism to other echinocandins but still being an orally administrated drug. IBX acts by inhibiting β-1,3-D-glucan synthase, a key enzyme in the biosynthesis of β-(1,3)-D-glucan, which is a major component of the fungal cell wall (Figure 2 and Figure 3).65
IBX is the first drug in the past 20 years to receive US Food and Drug Administration (FDA) approval (on 1 June 2021) for the treatment of VVC.66 Although IBX acts on the same enzyme as echinocandins, it has a different structure and interacts differently with the target enzyme, resulting in a lower rate of resistance.67–69 IBX is approved for the treatment of VVC in adult and post-menarchal paediatric females, with a recommended dose of 2 × 150 mg tablets administered approximately 12 hours apart for 1 day.
Spectrum of antifungal activity
Several studies investigating the activity of IBX have shown that it possesses fungicidal activity against Candida.70,71 This activity was reported to also include isolates that are resistant to azoles and echinocandins.69,71
IBX demonstrated potent activity against albicans and non-albicans Candida species reported to cause VVC,72 including C. glabrata – one of the fungi known to cause VVC, which recently began to exhibit increased resistance against commonly available antifungal agents.73 Schell et al. showed, using in vitro assays, that IBX maintains its activity against C.glabrata isolates that possess resistance against echinocandins.72 Similarly, IBX had MIC values ranging from <0.03 to 4 μg/mL against C.glabrata strains with mutations in the FKS gene (responsible for resistance against echinocandins).74 However, isolates with F641S, F649del, F658del and F659del mutations were less susceptible to IBX.68,69,74,75 Additionally, we previously demonstrated that among 22 echinocandin-resistant C. glabrata strains the minimum concentration of IBX that inhibits 50% of the tested isolates (MIC50) was 1 μg/mL and the minimum concentration of IBX that inhibits 90% of the tested isolates (MIC90) was 4 μg/mL.76 The MIC50 and MIC90 for caspofungin were 1 and 2 μg/mL, respectively, and micafungin demonstrated an MIC50 and MIC90 of 0.125 and 1 μg/mL, respectively.76
Pharmacokinetics
Several studies were performed to determine the pharmacokinetic parameters and bioavailability of IBX. In a phase I study conducted in healthy volunteers, IBX demonstrated a mean terminal half-life of approximately 20–30 hours. Additionally, IBX was shown to be safely administered with or without food, although a high-fat meal increased the bioavailability of IBX.77
In terms of tissue distribution, IBX displayed wide tissue distribution, which was several-fold greater than fluconazole and echinocandin.78 It achieved a steady-state volume of distribution of >5 L/kg in animal models.79 Furthermore, IBX demonstrated high tissue penetrability via its ability to achieve a kidney tissue concentration 20- to 25-fold greater than that seen in plasma. Moreover, a single oral dose of IBX (15 mg/kg) in rats, was sufficient to achieve a tissue-to-blood area under the curve (AUC) ratio of 9-fold in vaginal tissue.79 IBX showed a high potential to accumulate in vaginal tissue and fluids, with a tissue concentration 2-to 5-fold higher than that in plasma. Importantly, IBX was also shown to exhibit potent activity at a lower pH environment compared with azoles and echinocandins.56 In an in vitro study by Larkin et al., reduction of the pH resulted in increased activity of IBX, which was noted by the lower MIC90 value of <0.016 μg/mL at pH 4.5 versus 0.5 μg/mL at pH 7.56 In contrast, minimal or no change in the MIC values was observed with micafungin and fluconazole at a lower pH.56 This characteristic could be advantageous for IBX as the low vaginal pH will boost its activity, resulting in a better response to the treatment.
IBX had a half-life of approximately 20 hours and was eliminated mainly in the faeces.66 Following administration of carbon labelled [14C]-IBX intravenously and orally, approximately 90% of the radioactivity was recovered from faeces and bile for both administration routes.79 Similarly, in a separate study of six healthy human subjects, an average of 98% of [14C]-IBX administered orally was recovered in faeces.80
IBX is a CYP3A4 substrate and a reversible inhibitor of CYP2C8 and CYP3A4, as shown by in vitro studies. Despite this observation, co-administration of IBX with CYP2C8 and CYP3A4 substrates showed that IBX had minimal effects on the pharmacokinetics of both drugs. This suggests a low potential for IBX to cause CYP-mediated drug interactions at therapeutic exposures. In support of this concept, co-administration of a single dose of tacrolimus with IBX at a steady state resulted in a 1.4-fold increase in systemic exposure to tacrolimus with no effect on the maximum blood level, Cmax (maximum serum concentration that a drug achieves in a compartment of the body following administration of a single dose).81,82 Additionally, no age-dependent dose adjustment is required for IBX based on a study comparing the area under the curve from zero to infinity; the geometric mean ratio for total drug exposure across time (AUC0-inf) in elderly versus young healthy subjects (range of 65–76 years versus 20–45 years) was 1.39 (90% confidence interval 1.19, 1.62).83 Furthermore, IBX did not affect the AUC or Cmax levels of CYP2C8 substrates (e.g. rosiglitazone).82 Similarly, co-administration of IBX with a moderate CYP3A4 inhibitor (diltiazem) or proton pump inhibitor (pantoprazole) was not associated with clinically significant alteration in IBX AUC or Cmax levels.83 IBX did not significantly impact dabigatran, a substrate for P-glycoprotein, AUC or Cmax levels when both drugs were co-administered in healthy subjects.84
Taken together, these data suggest that, unlike azoles antifungals, IBX can be safely administrated with several other drugs without the need for dose adjustment.85 However, co-administration of IBX with strong CYP3A inhibitors or inducers should be done with caution as it may alter its plasma concentrations and, consequently, affect IBX safety and efficacy.83
Safety profile
IBX is designed to target β-1,3-D-glucan synthase, an enzyme unique to lower eukaryotes. Thus, IBX has a low risk of causing toxicity. In the VANISH 303 clinical trial that tested the safety of IBX versus placebo in patients with acute VVC, 39.7% (98/247) of the patients reported side effects compared with 16.9% (21/124) for the placebo-treated group.86 The majority of these side effects were gastrointestinal and mild in severity. The most highly reported side effects in the IBX-treated group were diarrhoea and nausea (22.3% [55/247] and 10.9% [27/247], respectively).86 Moreover, four patients in the IBX-treated group discontinued the study due to side effects. Interestingly, in the IBX-treated group, two pregnancies were reported, both of which resulted in a live birth with no associated delivery or newborn complications.86 However, in animal studies, oral administration of IBX in rabbits during organogenesis was associated with rare malformations such as absent forelimb, absent ear pinna, and thoracic gastroschisis at dose exposures greater or equal to approximately five times the human exposure at the recommended human dose. On the other hand, foetal toxicity was not observed in pregnant rats. Based on these data, IBX is contraindicated during pregnancy.83,87
In another clinical trial (VANISH 306), the safety of IBX versus placebo was evaluated in post-menarchal females aged ≥12 years with acute VVC.88 Of 298 patients in the IBX-treated group, 44 reported mild-to-moderate side effects, which were also limited to the gastrointestinal tract, versus 6 of 151 in the placebo-treated group.88 Additionally, only two patients discontinued the IBX treatment specifically due to abdominal pain and vomiting.
Similar results were observed in the clinical CANDLE trial that evaluated the safety of IBX versus placebo in patients with RVVC.89 In that study, 47 of 130 patients in the IBX-treated group reported gastrointestinal side effects compared with 21 of 130 of the placebo-treated group.89
Finally, unlike fluconazole, there was no clinically relevant effect on QTc interval. In this regard, the effect on QTc interval was tested on human subjects in which no significant change was observed with a plasma concentration up to 4 μg/mL.90 Thus, IBX, like other echinocandins, has a good safety profile. Clinical studies have only evaluated the safety and efficacy of IBX compared with placebo. A head-to-head comparison with fluconazole remains to be conducted.
Clinical development overview
Several clinical trials have been performed to test the efficacy and safety of IBX in the management of VVC. Ibrexafungerp-203, a multicentre, randomized, evaluator-blinded, active-controlled study to evaluate the safety and efficacy of oral IBX versus oral fluconazole in subjects with VVC was the first clinical trial conducted as proof-of-concept.91 This was followed by a phase II, multicentre, randomized, double-blind, double-dummy, active-controlled, dose-finding study to compare the efficacy, safety and tolerability of oral IBX versus oral fluconazole in adult female subjects 18 years and older with moderate-to-severe acute VVC (DOVE).92
Additionally, two phase III, multicentre, randomized, double-blind, placebo-controlled trials to evaluate the efficacy and safety of oral IBX versus placebo in subjects with acute VVC (VANISH) were conducted in which the IBX-treated group exhibited significantly higher rates of clinical cure (defined as complete resolution of all signs and symptoms of VVC) and mycological eradication compared with placebo (50.5% versus 28.6% and 49.5% versus 19.4%, respectively).93 IBX was shown to have a sustained effect in reducing VVC symptoms compared with placebo (59.6% versus 44.9%; p=0.009) at follow-up visit (~4 months after last dose). Additionally, the time to initial symptom relief ranged between 1 and 15 days with some cases reporting improvement as early as day 1. Moreover, the time to complete resolution of all symptoms ranged between 1 and 25 days. These effects were not altered by other factors such as race or high body mass index. Overall, IBX was well tolerated with minimal side effects primarily in the form of mild gastrointestinal symptoms, including vomiting and nausea.86,88,93
IBX has also been reported to effectively reduce the incidence of RVVC following the completion of a phase III, multicentre, randomized, double-blind, placebo-controlled study aimed to evaluate the efficacy and safety of oral IBX compared with placebo in female subjects 12 years and older with RVVC (CANDLE).89 The subjects in this study were given a once-a-month dose of IBX (300 mg twice a day) for prevention of RVVC for a total of 6 months. The primary endpoint in this study was clinical success, which was defined as subjects having a test-of-cure (TOC) evaluation with no mycologically proven, presumed or suspected recurrences of VVC up to TOC (week 24) visit. Mycologically proven recurrence was defined as an episode of VVC with a total composite score of ≥3 on the Vulvovaginal Signs and Symptoms (VSS) Scale and a culture-positive test for Candida spp. that required antifungal treatment. Presumed recurrence was defined as an episode of VVC with a total composite score of ≥3 on the VSS Scale that required antifungal treatment, and for which there was positive potassium hydroxide (KOH) microscopy, but no positive fungal culture. Suspected recurrence was defined as an episode of VVC that required treatment with an antifungal agent regardless of VSS composite score but for which there was no mycological evidence of disease.
Interestingly, in a study by Goje et al., 65% of the IBX-treated group achieved clinical success with no recurrence compared with 53% for the placebo-treated group.94 Additionally, no mycologically proven recurrence was reported in 70.8% of patients treated with IBX. The data from this study have been recently submitted to regulatory agencies for review, but approval of this new indication for prevention of RVVC has not been published as of the date of this paper.
Currently, a new clinical trial is being conducted to determine the potential use of IBX in treatment of patients with complicated VVC in whom prior fluconazole therapy has failed. In this study, three different IBX regimens are being tested: 1, 3 or 7 days of 300 mg IBX twice daily for a total of 600 mg per day. The study (NCT05399641) is currently still in the recruitment phase.95
Although IBX has been shown to have potent efficacy in the management of VVC compared with placebo, a head-to-head comparison with fluconazole has not been conducted. However, based on the available data, IBX can be used as an alternative to fluconazole for the management of VVC, especially in patients who have contraindication to fluconazole (e.g. allergic reaction) or those with infections resistant to fluconazole therapy.
Oteseconazole
Oteseconazole (previously VT-1161) is a new oral antifungal agent indicated to reduce the incidence of RVVC in females with a history of RVVC who are not of reproductive potential. In fact, it is the first antifungal agent to receive US FDA approval for this indication.96
According to the label, oteseconazole can be used alone or as a follow-up treatment after fluconazole. The single drug regimen consists of single dose of 600 mg, then 450 mg capsules of oteseconazole on day 1 and day 2, respectively, followed by 150 mg once a week for 11 weeks beginning on day 14. For the fluconazole–oteseconazole regimen, first fluconazole is initiated alone as a single oral dose of 150 mg every 72 hours, followed by 150 mg of oteseconazole once daily for 7 days from day 14 to 20, then 150 mg once a week for 11 weeks beginning on day 28.97
Although oteseconazole is a member of the azole antifungal group, its new chemical structure makes it different from the other agents. Currently available azoles contain an imidazole or triazole moiety that binds the human cytochrome P450 causing several side effects and drug–drug interactions, while oteseconazole contains a tetrazole moiety that increases the selectivity of oteseconazole for the fungal CYP51, reducing off-target interaction with the human cytochrome and consequently reducing side effects (Figure 3 and Figure 4).98,99
Spectrum of activity
Oteseconazole demonstrated a wide spectrum of activity against fungi isolated from patients with VVC comparable to or more potent than fluconazole. In a study performed by Ghannoum et al., 1,910 clinical isolates (87% C. albicans) were obtained from women with VVC, and the MIC50 and MIC90 for oteseconazole were reported to be 0.002 and 0.06 µg/mL, respectively, compared with 0.25 and 8 µg/mL for fluconazole, respectively.3 Furthermore, against C. glabrata, represented in 8% of these isolates, oteseconazole had MIC50 and MIC90 values of 0.03 and 0.125 µg/mL, respectively, compared with 2 and 8 µg/mL, for fluconazole, respectively.3
Similarly, in a study conducted by Wang et al., oteseconazole showed activity against C. glabrata, C. parapsilosis and C. tropicalis comparable to or more potent than that of itraconazole and fluconazole.100 The MIC50 and MIC90 for oteseconazole against C. glabrata were 0.5 and 2 µg/mL, compared with 1 and 2 µg/mL for itraconazole, and 2 and 64 µg/mL, for fluconazole, respectively. Against C. parapsilosis and C. tropicalis, oteseconazole had an MIC50 of 0.031 and 0.016 µg/mL compared with 0.125 and 0.5 µg/mL for itraconazole, and 0.25 µg/mL for fluconazole against both organisms, respectively. Additionally, while oteseconazole had an MIC90 similar to itraconazole against C. parapsilosis (1 µg/mL) compared with fluconazole, it was 4-fold dilution lower (i.e. 1 versus 16 µg/mL). A similar observation was also noted against C. tropicalis (1 versus 2 and >64.0 µg/mL, respectively).
These in vitro findings agree with in vivo data generated by Garvey et al. where 25 mg/kg oral doses of oteseconazole and fluconazole were sufficient to significantly reduce the fungal burden on day 1 post-treatment compared with the vehicle-treated group in a murine model of vaginal candidiasis infected with susceptible C. albicans strain.101 Although both drugs maintained their activity through 4 days post-treatment, oteseconazole showed superior activity against fluconazole-resistant strains using the same animal model. However, against one strain that was moderately resistant to oteseconazole, no significant effect was observed with both antifungals.101
A study investigating the resistance mechanism of oteseconazole showed that overexpression or mutation of the active site (e.g. mutations in ERG3) of the fungal lanosterol 14α-demethylase was associated with reduced susceptibility to this agent.102 Additionally, upregulation of the drug efflux pumps was another mechanism that was associated with resistance to oteseconazole.
Taken together, oteseconazole has demonstrated high activity against a wide variety of Candida isolates including azole-resistant strains. However, taking into consideration our experience with fluconazole, it will be important to practice antimicrobial stewardship to avoid development of resistance.
Pharmacokinetics
In clinical trials evaluating the pharmacokinetics of oteseconazole, the plasma concentration of the drug was shown to be increased in a dose-dependent manner. Following oral administration, around 76% of the antifungal agent was absorbed. Additionally, approximately 5–10 hours were needed for oteseconazole to reach the peak plasma concentration achieving a Cmax of 2.8 ± 1.25 following repeat dose administration. However, the Cmax value was increased by 45% following oteseconazole intake with a high fat meal compared with fasting state.97
Oteseconazole is highly bound to plasma protein (99.5–99.7%) with a volume of distribution of 423 L. Additionally, data from animal studies showed that oteseconazole achieved a vaginal concentration 2-fold higher than that in the plasma following oral intake.101 Oteseconazole was also shown to have a low rate of metabolism, with a median terminal half-life of approximately 138 days. Moreover, it is mainly excreted in faeces and urine (56% and 26%, respectively).
In terms of drug interaction, oteseconazole increased the Cmax and area under the curve from zero to 24 hours (AUC0-24h) of rosuvastatin, a breast cancer resistance protein (BCRP) substrate, by 118% and 114%, respectively. This may increase the incidence of BCRP substrate side effects. Thus, co-administering oteseconazole with a BCRP substrate requires starting the BCRP agent at the lowest possible dose and monitoring for development of side effects. On the other hand, no clinically significant differences in the pharmacokinetics of the following drugs were observed when co-administered with oteseconazole: midazolam (sensitive CYP3A4 substrate), ethinyl estradiol (CYP3A4 substrate), norethindrone (CYP3A4 substrate) or digoxin (P-gp substrate).
Safety profile
Because of the unique structure of oteseconazole (having tetrazole moiety) as mentioned above, it has high selectivity for the fungal CYP51 with reduced affinity for the human type.100 Consequently, oteseconazole was shown to have fewer side effects compared with other members of the azole antifungals, with the most common side effects being headache and nausea.103,104
Oteseconazole is contraindicated in females of reproductive age, in pregnant women and during lactation.97 This warning, indicated in the drug label, was based on animal studies in which administration of oteseconazole to pregnant rats and rabbits resulted in ocular abnormalities. Furthermore, considering the drug exposure window of approximately 690 days, reducing the embryo-foetal toxicity risks is challenging.97
In patients with mild renal or liver diseases, dose adjustment is not required. However, due to insufficient numbers of subjects evaluated belonging to these populations, oteseconazole is not recommended in severe disease.97
Clinical development overview
Three phase III clinical trials have been conducted for evaluating oteseconazole.103–105 In the first two trials, C. albicans represented 87% of the organisms isolated from the vaginal swabs followed by C. glabrata which was the causative organism in 8% of the cases. Other organisms that were identified included: C. parapsilosis, C. tropicalis, C. krusei, C. dubliniensis, C. kefyr and Saccharomyces cerevisiae.
Participants with VVC were treated with one dose of fluconazole 150 mg every 72 hours, and those with resolved symptoms and signs were randomly assigned to a treatment group in a ratio of 2:1 to receive 150 mg of oral oteseconazole daily for 7 days, followed by 150 mg of oteseconazole weekly for 11 weeks, or to matching placebo for 12 weeks. Patients in both groups were then followed up with no treatment for 36 weeks.
The primary endpoint in both studies (CL-011 and CL-012) was the proportion of patients with ≥1 culture-verified acute VVC episode during the maintenance phase. The incidence in the oteseconazole-treated group ranged between 3.9% and 6.7% versus 39.0% and 42.8% for the placebo-treated group. When considering recurrence – patients with culture-verified VVC or those who received an antifungal treatment for a VVC episode during the study – the reported incidence of recurrence ranged from 21.3% to 27.3% for the oteseconazole group and 49.7% to 50.8% for the placebo group. These analyses were performed using missing values with multiple imputation. Additionally, treating culture-proven recurrences, VVC medication use, and incomplete follow-up as failures led to a 35% to 42% failure rate for oteseconazole versus 56.5% to 57.8% for placebo, maintaining a 14.9% to 21.7% difference between the groups and realigning statistical significance for this more strict analysis.105
The percentage of side effects were similar between the treatment groups, with the most common side effects being respiratory infections, headache, bacterial vaginosis, vulvovaginal pruritus, urinary tract infection, cystitis and back pain.105 Interestingly, no adverse obstetric reactions or foetal abnormalities were reported among eight pregnant participants who were in the oteseconazole treatment group, with six participants giving birth to healthy babies, one participant electively terminating the pregnancy, and one with an unknown outcome.
Similarly, in the ultraVIOLET clinical trial that included patients with a history of RVVC with a confirmed acute VVC episode at the time of screening, the most common organisms isolated from patients’ samples were C. albicans (76.1%), C. glabrata (11.8%), C. parapsilosis (5.4%) and C. tropicalis (4.1%).104
The primary efficacy outcome measure was the proportion of participants with ≥1 culture-verified acute VVC episode during the maintenance phase of the study through week 50 in the intent-to-treat (ITT) population (including all randomized participants, inclusive of those with unresolved infection during the induction phase). Multiple endpoints were used as secondary outcomes, including: 1. the proportion of participants with resolved acute VVC infection at the end of the induction phase; 2. the proportion of participants with ≥1 culture-verified acute VVC episode with a signs and symptoms score of ≥3 during the maintenance phase; 3. time to first recurrence of culture-verified acute VVC episode with a signs and symptoms score of ≥3 during the maintenance phase; and 4. proportion of participants with ≥1 positive culture for Candida during the maintenance phase.
The results of the primary efficacy outcome showed that only 5.1% of the oteseconazole-treated group had ≥1 culture-verified acute VVC episode compared with 42.2% of the group receiving placebo through week 50 post-randomization (p<0.001). When use of VVC medication was considered as part of the definition of recurrence, the recurrence rate of the oteseconazole group was 43.5% versus 59.0% of the fluconazole/placebo group (p=0.039). Furthermore, when considering recurrences, VVC medication use and incomplete follow-up as failures by definition, the recurrence was 59.2% for the oteseconazole group compared with 70.8% for the fluconazole/placebo group, which supports the efficacy of oteseconazole compared with fluconazole/placebo in reduction of recurrence. This analysis was performed using multiple imputation to address subjects with incomplete data.
In the induction phase (treatment of cases with acute VVC), oteseconazole was similar to fluconazole in the treatment of acute VVC, with 93.2% of the oteseconazole-treated group reporting resolution of symptoms compared with 95.8% in the fluconazole-treated group at day 14. Additionally, 34.0% (50/147) of the oteseconazole-treated group reported failure to resolve by the end of the induction phase (day 14) or recurrence during the maintenance phase compared with 44.4% (32/72) for fluconazole/placebo group. When incomplete follow-up was treated as a failure, the percentages were 51.0% for oteseconazole compared with 66.7% for fluconazole/placebo (p=0.028).
No significant differences were noted between the two treatment groups in terms of side effects (54% and 64% for oteseconazole- and fluconazole/placebo-treated groups, respectively). In line with the results obtained from previous trials, the most common side effects were infections (36% versus 44%, respectively), mostly in the form of urinary tract infection and bacterial vaginosis, and gastrointestinal disorders (10% versus 11%, respectively).104
Future directions
Although we currently have two new options for the management of VVC and RVVC, there is a need to develop more options that can meet patients’ needs and complement the available therapies. In this regard, several trials are ongoing to evaluate new potential therapies. Beside agents that act through their antimicrobial activity, several options are being developed to target other aspects of pathogenesis such as preventing Candida colonization, enhancing the immune response and rebalancing the vaginal microbiome. In this section, we will briefly discuss potential candidates under development to treat VVC.
Vaccines against vulvovaginal candidiasis currently in clinical trials
NDV-3A vaccine
The development of an effective, long-lasting and safe vaccine could be a key to lowering the incidence of VVC. N acetyl delivery vaccine-3 (NDV-3) comprises a recombinant agglutinin-like sequence 3 protein (Als3p) epitope in an adjuvant delivery system. The suggested mechanism of action is that NDV-3 works by triggering both humoral and adaptive immune responses by producing long-lasting memory B- (immunoglobulin [Ig]G, IgA1) and T-cell (interferon-a, interleukin-17A) responses after the first dose, which will be boosted with a second dose.106 In a clinical trial by Edwards et al., the NDV-3A vaccine was highly immunogenic, safe and decreased the frequency of VVC symptomatic episodes in women aged <40 years for up to 12 months.107 However, this was not the case for patients older than 40 years old. The impact of the vaccine in older patients was weaker compared with those aged <40 years. This may limit the utility of the vaccine as it will not provide appropriate coverage for a large portion of VVC patients.
PEV7 vaccine
The virosome formulated anti-Candida vaccine (PEV7) is a shortened recombinant secretory aspartyl proteinase 2 (rSAP2) protein integrated into influenza virosomes that has demonstrated some protection against VVC in rats.108 Following intravaginal or intramuscular plus intravaginal administration, high levels of anti-Sap2 IgG and IgA were found in the vaginal fluid of rats. PEV7 was also found to be safe in repeated-dose toxicological testing in rats. The vaccine is currently undergoing a human trial (phase I) by Pevion Biotech AG.108
Whilst overall results are promising, it is hypothesized that simultaneously targeting numerous Candida epitopes (multivalence) will probably be more effective in eliciting a stronger and wider immune response, as Candida antigens are complex and have the capacity to evade host immune surveillance. This will provide a future direction for vaccine development against vaginal yeast infections worldwide.109,110 However, no clinical trials or studies are in progress regarding this vaccine approach.
Approaches to rebalance the microbiome
Probiotics
As VVC is associated with alterations in the abundance of Lactobacillus species in the vaginal milieu, efforts to use probiotics to rebalance the vaginal microbial communities are underway.14–16 The mechanism by which probiotics may exhibit their effect is by positively modulating the vaginal microbiota to increase beneficial microorganisms, thereby preventing candidal overgrowth.
Data reported from different studies are variable regarding the efficacy of probiotics in management of VVC. Where some studies reported successful utilization of probiotics for management of VVC,111–113 others failed to demonstrate significant difference between the standard VVC treatment alone versus when combined with probiotics.114,115 For example, the study by Oerlemans et al., evaluated the impact of a probiotic vaginal gel containing three species of Lactobacilli (Lactobacillus rhamnosus GG, Lactobacillus pentosus KCA1 and Lactobacillus plantarum WCFS1) on VVC.116 Data analysis showed that 45% of women did not need rescue treatment, suggesting an improvement in their symptoms.116 In another trial that included 95 patients, no significant difference in the clinical cure rate was noted between subjects who received probiotic capsules (containing Lactobacillus gasseri LN40, Lactobacillus fermentum LN99, Lactobacillus casei subsp., Lactobacillus rhamnosus LN113 and Pediococcus acidilactici LN23) and those receiving placebo treatment.115 Thus, additional studies evaluating the utility of probiotics to treat VVC are warranted.
Other alternative modalities under investigation for the management of vulvovaginal candidiasis
TOL-463
TOL-463 is a non-azole, lactobacilli-sparing, boric acid-based vaginal anti-infective enhanced with edetic acid. A study by Marrazzo et al. evaluated this formulation in a randomized, single-blind, phase II, controlled clinical trial and showed that TOL-463 was safe and well tolerated in the treatment of bacterial vaginosis and VVC. However, vulvovaginal burning was the most frequently encountered adverse event, which may limit patient compliance.117
Dequalinium chloride (Fluomizin®)
Dequalinium chloride (DQC; Fluomizin; Kora Healthcare, Dublin, Ireland) is a quaternary ammonium salt with established antimicrobial activity against a wide range of vaginal pathogens, including bacteria, Candida species (e.g. C. albicans and C. glabrata) and Trichomonas vaginalis.118,119 It acts primarily by increasing the microbial cell permeability followed by the loss of mitochondrial ATP synthesis via inhibition of F1-ATPase, leading to cell death.120 DQC demonstrated rapid in vitro bactericidal and fungicidal effects after a short contact time (within 30–60 minutes). Additionally, when compared with clotrimazole, both medications showed comparable clinical response; however, the latter resulted in higher negative KOH microscopic examination and fungal culture.121,122
Blue light-emitting diode
The antimicrobial effect of blue light-emitting diodes (LEDs) make it a good potential candidate for the treatment of VVC. It acts by targeting the endogenous porphyrin of the pathogens resulting in cell death. Preliminary results of an ongoing clinical trial using 401 ± 5 nm blue LED demonstrated that it did not affect the microbiota in any of the participants, albeit with no adverse effects observed during or after illumination.123 The next step is to test the effect of 401 ± 5 nm blue LED on pathogens causing vulvovaginal infection. This approach is still in its infancy.
Medical-grade honey formulation (L-Mesitran®)
L-Mesitran (Triticum Exploitatie BV, Maastricht, The Netherlands) is an organic honey that is sterilized with gamma radiation, and it has been used as a potential antifungal treatment. A trial investigating the effect of L-Mesitran administration on RVVC symptoms is currently recruiting.124
Conclusion
VVC represents a disabling disease125 that affects the quality of life of women, and development of antifungals to treat this disease is lacking. Fortunately, recently, two new and effective antifungals that target VVC and RVVC were approved by the US FDA. Moreover, it is encouraging that alternative approaches to managing this disease are currently being pursued.