Heavily treatment-experienced people living with HIV (HTE-PLWH) represent a minority of those living with HIV.1 While there is variability in the definitions of HTE-PLWH, its prevalence is estimated to be between 1 and 10% of the total number of people living with HIV (PLWH) monitored in high-income countries.2,3
HTE-PLWH are at high risk for clinical progression, virologic failure and death.4,5 However, few, if any, treatment options are available for HTE-PLWH due to their archived drug resistance mutations and/or intolerance to the therapy itself.6 Even those with controlled viraemia often face extremely complex regimens with multiple drugs and different modes of administration (e.g. once or twice a day or via oral, subcutaneous or intravenous administration).1 Therefore, new classes of drugs with long half-lives allowing for long-acting administration are desirable in this population, as substantial problems with antiretroviral therapy (ART) adherence often occur.7
Lenacapavir (LEN; Gilead Sciences, Inc., Foster City, CA, USA) is a first-in-class HIV capsid (CA) inhibitor with pharmacokinetic properties that allows for subcutaneous dosing every 6 months.8,9 The pivotal phase II/III CAPELLA trial (ClinicalTrials.gov identifier: NCT04150068),10,11 has demonstrated its efficacy, even in participants with no active drug in their optimized background regimen (OBR). Based on the results from the CAPELLA trial, LEN has been approved by both the US Food and Drug Administration (22 December 2022) and the European Medicines Agency (25 August 2022) for the treatment of HTE-PLWH with a multidrug-resistant virus that cannot be successfully treated with other available treatments due to resistance, intolerance or safety considerations.12,13 In addition, LEN has been investigated for use in PLWH who are naïve to ART and as a possible pre-exposure HIV prophylaxis in the phase II clinical trial CALIBRATE (ClinicalTrials.gov identifier: NCT04143594) and the phase III PURPOSE 1 (ClinicalTrials.gov identifier: NCT04994509) and PURPOSE 2 (ClinicalTrials.gov identifier: NCT04925752) clinical trials.14,15
This review summarizes and discusses the main aspects of LEN, including its mechanism of action, pharmacokinetic/pharmacodynamic properties, and clinical efficacy and safety findings. Finally, we will discuss the potential implications of LEN approval for the management of HTE-PLWH.
Mechanism of action of lenacapavir
The HIV CA contributes to various aspects of the replication cycle of HIV-1 at both the early and late stages of the viral life cycle.16,17 In particular, the CA protects the viral genome after cell entry and favours reverse transcription of the viral RNA genome into double-stranded DNA, thus creating a confined environment with a high concentration of viral components; through its disassembly, the CA allows for the release of genetic material into the cell nucleus, thereby supporting the subsequent integration of proviral DNA.9,18 The CA contains ~1,500 proteins arranged in approximately 250 hexamers and 12 pentamers, each comprising monomeric CA proteins. Upon cleavage by HIV-1 proteases, CA proteins self-assemble into a conical CA, the correct formation and integrity of which is essential for viral infectivity.9,19
LEN is a small molecule that blocks multiple steps in the HIV-1 life cycle, including CA-mediated nuclear uptake of HIV-1 proviral DNA, by blocking nuclear import proteins from binding to the CA; virus assembly and release by interfering with Gag/Gag-Pol function and reducing CA subunit production; and CA core formation by disrupting the rate of subunit association resulting in misshapen tubular CAs rather than the functional conical shape.9,16,17,20 With respect to CA core formation, LEN tightly binds to a conserved interface between CA protein monomers. This binding specifically blocks the formation of CA pentamers but not hexamers.19 Consequently, LEN treatments result in open-ended tubular assemblies containing only hexamers. The absence of pentamers inhibits the formation of a properly structured CA.
Pharmacokinetics
Following subcutaneous administration, LEN is gradually released and fully absorbed.9,20,21 Absolute bioavailability following oral administration is low, varying between 6% and 10%. Time to peak drug concentration occurs at about 4 hours following oral administration and 84 days following subcutaneous administration.
The oral and subcutaneous administration of LEN have median half-lives of 10–12 days and 8–12 weeks, respectively.20,21 The pharmacokinetics of oral LEN does not appear to be affected by food.20,21 Until 28 weeks post-subcutaneous administration, the LEN trough concentration is safely kept above the efficacy target, which allows for subcutaneous dosing of LEN every 6 months (26 weeks) with a 2-week dosing window.22 People who are unable to receive LEN subcutaneously during this period (i.e. those whose dose exceeds the 28-week window) need to restart an oral LEN loading dose followed by subcutaneous LEN.22
In vitro, 99.8% of LEN is bound to plasma proteins, highlighting the drug’s high protein binding rate.20,21 Hence, dialysis should not affect LEN exposure.20,21
The main elimination process of LEN is the excretion of unchanged drug in the faeces.20,21 Although to a minor extent, LEN metabolism is primarily mediated by cytochrome P450 3A4 (CYP3A4) and UDP glucuronosyltransferase family 1 member A1 (UGT1A1). However, as no single circulating metabolite accounts for more than 10% of drug-related plasma exposure, LEN metabolites have not been thoroughly characterized.20,21
In healthy volunteers who received a single intravenous dose of radiolabelled LEN, 76% of the total radioactivity was recovered from faeces samples and <1% from urine samples.21 Unchanged LEN was predominately found in plasma (69%) and faeces (33%) samples.20,21 Participants with severe renal impairment (estimated creatinine clearance value of 15–29 mL/min) and moderate-to-severe liver disease have a higher LEN exposure, which is assessed using plasma concentrations, than healthy subjects; however, these increases are not considered clinically significant.21,23,24
Data are lacking for the use of LEN in pregnant women. However, neither pregnancy, nor parturition, nor the postnatal period have been shown to be negatively affected by LEN exposure in animal studies.20 Currently, the use of LEN during pregnancy should only be considered if it is necessary to treat clinical conditions.20 Although it is unknown whether LEN is excreted in human milk, LEN has been found in the plasma of nursing rat pups during animal studies, with no associated clinical events.20
Antiviral activity in vitro
In vitro data from HIV-1-infected cells have shown that LEN has half-maximal effective concentrations (EC50) of 105 pmol/L in MT-4 cell lines, 32 pmol/L in primary human CD4+ cells and 56 pmol/L in macrophages.9,16,17 LEN has also demonstrated antiviral activity against all major HIV-1 subtypes, including A, A1, AE, AG, B, BF, C, D, E, F, G and H.9,16
Due to the fact that HIV-2 is less common and virulent than HIV-1, fewer data are available to study the effectiveness of antiretroviral (ARV) drugs. However, recent data have shown that LEN potency against HIV-2 is reduced 11- to 16-fold compared with HIV-1, regardless of the presence of drug resistance mutations in HIV-2 reverse transcriptase (RT) or integrase.25 Given this difference in potency, treatment of PLWH-2 with a LEN-based regimen may require careful monitoring to assess virologic response. Despite the lower potency, the potential clinical utility of LEN in HTE-PLWH-2 may not be impaired. However, the in vivo data are lacking.25
Clinical efficacy data in heavily treatment-experienced individuals
The phase II/III CAPELLA trial
The on-going phase II/III CAPELLA study (ClinicalTrials.gov identifier: NCT04150068) is evaluating the efficacy and safety of LEN in HTE-PLWH.10,11,26 The CAPELLA study enrolled participants with stable but ineffective ART (HIV-1 RNA ≥400 copies/mL) who had documented resistance to at least two ARV drugs from at least three of the four main classes (nucleoside RT inhibitors, non-nucleoside RT inhibitors, integrase strand transfer inhibitors and protease inhibitors), and no more than two fully active ARV drugs from the four main classes that could be effectively combined.
Changes in plasma HIV-1 RNA levels between the screening and cohort selection visits, which occurred 14–30 days apart, were used to divide participants into two distinct cohorts. Cohort 1 consisted of the first 36 participants with an HIV-1 RNA level of ≥400 copies/mL and a decline of <0.5 log10 copies/mL between the screening and cohort selection visits. In this cohort, participants were randomly assigned in a 2:1 ratio to receive oral LEN (600 mg on days 1 and 2 and 300 mg on day 8) or a matching placebo while continuing their failing therapy (double-blind). In the maintenance phase, which started on day 15, participants in the LEN group received subcutaneous LEN (927 mg) once every 6 months plus OBR. Participants in the placebo group received oral LEN (600 mg on days 15 and 16 and 300 mg on day 22), followed by subcutaneous LEN plus OBR.
Cohort 2 was designed to include participants with a decrease of ≥0.5 log10 copies/mL between screening and cohort selection visits, a viral load of ≤400 copies/mL or both. All participants in this cohort received open-label oral LEN (600 mg on days 1 and 2 and 300 mg on day 8) with OBR on day 1, followed by subcutaneous LEN every 6 months starting on day 15.
Primary and secondary efficacy endpoints were evaluated in cohort 1. The primary efficacy endpoint was the proportion of participants with a decrease in plasma HIV-1 RNA of at least 0.5 log10 copies/mL from baseline by day 15 (the end of the functional monotherapy). Secondary endpoints were the proportion of participants with a viral load of <50 copies/mL and the proportion of those with a viral load of <200 copies/mL at week 26 following initiation of subcutaneous LEN. Other key efficacy endpoints included changes in viral load and CD4+ counts.
A total of 72 participants were enrolled in the CAPELLA study. Of these, 36 were enrolled in cohort 1 (12 of whom were assigned to receive placebo during the functional monotherapy period and 24 to receive LEN), and 36 were enrolled in cohort 2. Participants in cohort 1 had received a median of 9 ARV treatments prior to enrollment, and their median overall susceptibility score for failing regimens was 0.8.10 The drug susceptibility score for an individual ARV drug was calculated using an algorithm that assigned a value of 1.0 for full susceptibility, 0.5 for partial susceptibility and 0.0 for no susceptibility; the overall susceptibility score was the sum of the individual susceptibility scores.
Participants in cohort 2 had similar characteristics (e.g. demographics, characteristics of HIV infection, viral resistance and immunovirological profiles) to participants in cohort 1.
In cohort 1, the primary efficacy endpoint (-0.5 log10 copies/mL HIV-1 RNA at day 15) was achieved in 21/24 (88%) participants in the LEN group and 2/12 (17%) participants in the placebo group during the functional monotherapy period (absolute difference 71%, 95% confidence interval [CI] 35–90; p<0. 001). At week 26, 29/36 participants from this cohort had HIV-1 RNA <50 copies/mL (81%;95% CI 64–92), and 32/36 participants had <200 copies/mL (89%; 95% CI 74–97. In cohort 2, 30/36 (83%) participants and 31/36 (86%) participants had viral loads of <50 copies/mL and <200 copies/mL, respectively, at week 26.
When considering both the randomized and non-randomized cohorts, 56/72 (78%) participants had a viral load of <50 copies/mL, 11/72 (15%) had HIV-1 RNA levels of >50 copies/mL and 5/72 (7%) had no virologic data at week 52.11 Of the 72 participants, 59 (82%) had a 52-week viral load of <200 copies/mL, 8 (11%) had an HIV-1 RNA level of >200 copies/mL and 5 (7%) had no virologic data. At week 52, 27/34 (79%) participants treated with two active ARVs in combination with LEN had a viral load of <50 copies/mL, while 20/26 (77%) participants with only one active ARV had a viral load of <50 copies/mL and 9/12 (75%) with no active ARV in combination with LEN had a viral load of <50 copies/mL. In addition, efficacy at week 52 was similar in different subgroups according to demographic characteristics (e.g. sex at birth, age and race), baseline CD4 count and HIV-1 RNA levels (although a trend toward a higher rate of virologic response was seen in those with lower viraemia and a higher CD4 cell count), and OBR characteristics.27
In terms of safety, a combined analysis of cohorts 1 and 2 at week 26 showed that 7/72 (10%) participants experienced serious adverse events (AEs), none of which were considered to be related to LEN by the investigator.10 After excluding injection site reactions (ISRs), the most common AEs were diarrhoea (11%) and constipation (11%). A total of 45/72 (63%) participants experienced at least one ISR. The ISRs included pain (31%), swelling (31%), erythema (25%) and nodule occurrence (24%). Most ISRs, including pain, were grade 1 and resolved within a few days. No grade 4 ISRs were reported.
After 52 weeks of follow-up, no study drug-related AEs occurred in >5% of participants, and no study drug-related serious AEs occurred.11 Three-quarters or more of the study participants had no ISRs after the first and second subcutaneous doses of LEN, and no grade 4 ISRs were reported. The three grade 3 reactions were swelling and erythema in one participant, which resolved in 4 and 8 days, respectively, and pain in another participant, which resolved in 1 day.11 Two participants died during follow-up: one from non-Hodgkin’s lymphoma and the other from acute respiratory failure; neither death was related to LEN.10,11
The efficacy of LEN was also evaluated in treatment-naïve PLWH in the CALIBRATE study (ClinicalTrials.gov identifier: NCT04143594).14,28,29 In this study, participants were randomized into four groups: group 1 and group 2 received 927 mg subcutaneous LEN every 26 weeks (after 2 weeks of oral LEN dosing) with oral tenofovir alafenamide/emtricitabine for the first 28 weeks of the study, then switched to oral tenofovir alafenamide (group 1) or bictegravir (BIC) (group 2); group 3 received oral daily LEN with emtricitabine and tenofovir alafenamide; and group 4 received oral daily BIC, emtricitabine and tenofovir alafenamide. Subcutaneous LEN showed good overall efficacy in groups 1 and 2. At week 54, an HIV-1 RNA level of <50 copies/mL was achieved in 47/52 (90%) participants in group 1 (difference with oral tenofovir alafenamide/emtricitabine and BIC comparator arm -2.6%, 95% CI -18.4 to 13.2) and 45/53 (85%) participants in group 2 (difference with the tenofovir alafenamide/emtricitabine/BIC comparator arm -7.1%, 95% CI -23.4 to 9.3).14 At week 80, the proportion of participants with viral suppression was 85% and 75% in groups 1 and 2, respectively, compared with 92% in the tenofovir alafenamide/emtricitabine and BIC control arm.28
The phase Ib trial of teropavimab and zinlirvimab in combination with lenacapavir
According to the pharmacokinetic characteristics of the subcutaneous formulation of LEN, combining LEN with other drugs characterized by long half-lives could allow to design ARV regimens with 6-monthly dosing that could benefit HTE-PLWH. In a phase Ib clinical trial (ClinicalTrials.gov identifier: NCT04811040), Eron et al. evaluated a regimen of LEN and two broadly neutralizing antibodies, teropavimab and zinlirvimab (Gilead Sciences, Inc., Foster City, CA, USA), administered every 6 months in ART-experienced PLWH with viral suppression.30 Teropavimab is a broadly neutralizing antibody that targets the CD4 binding site on HIV gp120, while zinlirvimab is a broadly neutralizing antibody that targets a non-overlapping epitope of the V3 glycan of HIV envelope glycoprotein. Both antibodies have been modified to extend their half-life and allow for 6-month dosing.
This phase Ib clinical trial enrolled 21 PLWH on ART with viral suppression for at least 18 months, with a baseline CD4+ count of > 500 cells/mm3 and a nadir CD4+ count of <350 cells/mm3.30 All study participants had a virus that was susceptible to both antibodies. At baseline, active ART was stopped, and all participants received an oral loading dose of 600 mg of LEN (repeated on day 2). Participants received two subcutaneous injections for a total of 927 mg of LEN and an intravenous infusion of teropavimab (30 mg/kg). They were also randomized to receive either 10 mg/kg or 30 mg/kg of zinlirvimab. In both groups, 10 participants received all scheduled doses and were included in the analysis. Although the study was originally designed to last 52 weeks, it was shortened to 26 weeks due to a clinical hold on the subcutaneous LEN formulation.
LEN, teropavimab and zinlirvimab (at both doses) remained above therapeutic levels (5 ng/mL for LEN and 2 μg/mL for teropavimab and zinlirvimab) for up to 26 weeks. At the end of the study, 90% of participants in both groups maintained viral suppression. One participant in the 30 mg/kg zinlirvimab group withdrew from the study at week 12. Another participant in the 10 mg/kg group experienced a viral rebound, which was suppressed after restarting the baseline regimen. Treatment was safe and generally well tolerated. No serious or life-threatening AEs or clinically significant laboratory abnormalities were observed, and there were no discontinuations due to AEs. The most common AEs were ISRs.30
Mutations associated with resistance to lenacapavir
In vitro
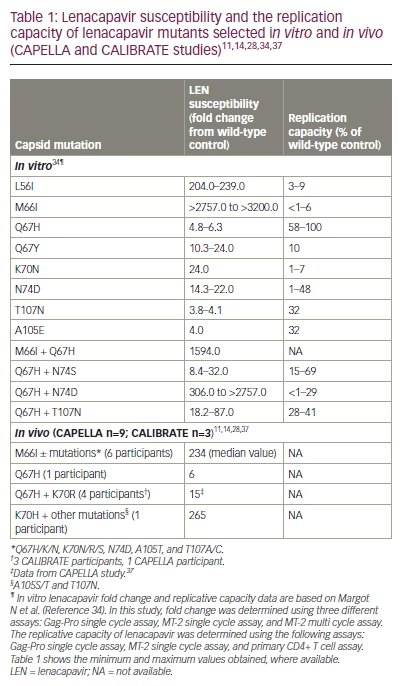
In vitro resistance selection assays have shown that Q67H and N74D are the main resistance-associated mutations (RAMs) in the gag gene associated with LEN exposure.9,31–33 Additional mutations include L56I, M66I, K70N, Q67H/N74S, and Q67H/T107N.9,31–33 These mutations, alone or in combination, confer reduced susceptibility to LEN (6- to >3,200-fold resistance compared with the wild type). Moreover, all but the low-level resistant variant Q67H (6-fold resistance to LEN relative to the wild-type virus) have been associated with a reduced replication capacity in vitro.34
LEN retains potent antiviral activity against HIV-1 site-directed mutants and clinical isolates resistant to currently approved ARV agents, including nucleoside RT inhibitors, non-nucleoside RT inhibitors, integrase strand transfer inhibitors, protease inhibitors, entry inhibitors (fostemsavir, ibalizumab and maraviroc) and the experimental drug islatravir (Merck & Co., Inc., Rahway, NJ USA).32,35,36 Due to its first-in-class nature, LEN is expected to be fully active regardless of the patient’s treatment history. In a sample of 1,500 PLWH, including treatment-naïve and treatment-experienced individuals, none of the LEN resistance mutations identified during the in vitro selection experiments were detected.31
In vivo
In this section, we present resistance data from the CAPELLA and CALIBRATE trials, which assessed the efficacy and safety of LEN in HTE- and treatment-naïve PLWH, respectively.10,11,14,28,37 The analysis of potential treatment-emergent resistance to LEN in the CAPELLA trial was carried out during the study’s maintenance phase when all participants received LEN plus OBR. Participants were tested for genotypic and phenotypic resistance to LEN and OBR components in the event of virological failure.10 By week 52, 21/72 (29%) participants met the criteria for resistance analysis, 9/72 (13%) developed LEN RAMs in the CA, and 12/72 (17%) did not meet the criteria for resistance analysis. Four major patterns of LEN RAMs were observed, including M66I ± other substitutions were the most common patterns, with 6/72 (8%) participants attesting M66I mutations and a median LEN phenotypic fold change (FC) of 234 in patients with M66I mutations compared with the wild type; the Q67H + K70R combination was found in 1/72 (1%) participants and was associated with a LEN FC of 15 compared with the wild type; and a K70H mutation was found in 1/72 (1%) participants and was associated with a LEN FC of 265 compared with the wild type.37 Eventually, the isolated LEN Q67H mutation emerged in a single participant at week 52, with an associated LEN FC of 6 compared to wild type.11 No participant with LEN resistance experienced the emergence of additional RAMs to the components of OBR.
Of the participants who did not develop resistance to LEN, 3/12 (25%) remained viraemic throughout the study without acquiring new OBR resistance. Ultimately, 9/12 (75%) participants suppressed their HIV-1 RNA level to <50 copies/mL without changing OBR, including two who initially experienced RAMs on their OBR.10 Of the 9 participants who developed resistance, 4 (44%) with LEN-associated CA RAM emergence were on functional LEN monotherapy and did not have fully active ARVs on their OBR.11 These participants had few treatment options based on baseline resistance analyses; however, two of them were able to resuppress their HIV-1 RNA level to <50 copies/mL after switching to active or partially active agents while maintaining LEN, and two participants experienced viral rebound to levels similar to baseline. The other five participants who developed LEN RAMs received at least two fully active OBRs. However, blood samples obtained during the development of LEN resistance revealed undetectable levels of several OBR drugs (darunavir, dolutegravir, emtricitabine and tenofovir), indicating poor adherence to oral ART.10,11
In the CALIBRATE study, three treatment-naïve PLWH developed resistance.14,28 At week 10 (during treatment with tenofovir alafenamide/emtricitabine plus subcutaneous LEN), the LEN-associated Q67H and K70R CA substitutions and the M184M/I RT mutation were detected in the first participant. Emtricitabine and tenofovir concentrations were consistent with expected pharmacokinetics, and LEN plasma concentrations were within target ranges (>3.87 ng/mL).38 At week 54, a second participant treated with emtricitabine/tenofovir alafenamide + oral LEN (group 3) experienced virologic rebound with the emergence of LEN resistance (Q67H mutation) and the subsequent emergence of the K70R mutation.28 Finally, a third participant receiving oral tenofovir alafenamide + subcutaneous LEN (group 1) developed the Q67H + K70R combination at week 80.28 A summary of the LEN RAMs selected in vitro and in vivo from the CAPELLA and CALIBRATE studies is provided in Table 1.11,14,28,34,37
Role of lenacapavir in the management of eavily treatment-experienced people living with HIV
The current article reviewed results from on-going clinical trials that, despite the limited sample size, have shown the efficacy and safety of LEN in HTE-PLWH. In the CAPELLA trial, virologic efficacy of 78% was achieved at 52 weeks, with a limited proportion of participants experiencing grade 3–4 AEs.10,11 In addition, ISRs, albeit frequent, were mostly mild to moderate in intensity.10,11
The long-acting, subcutaneous LEN formulation, which can be administered every 6 months, is a significant addition to the HIV treatment armamentarium due to its unique characteristics. However, despite data demonstrating the efficacy of regimens containing LEN as the only active drug, LEN should not be misused in the HTE population.11 Although it is often difficult to design regimens with at least two active drugs for HTE patients with failing ART, the add-on strategy (where an active drug is added to a failing regimen) should be strongly discouraged for LEN.6,39 Fortunately, new therapies from new drug classes have become available in recent years (e.g. fostemsavir and ibalizumab) or may be available soon (islatravir and broadly neutralizing antibodies).7,40 Consequently, LEN should be combined with an OBR that includes, when feasible, at least a second active agent that takes into account both new and existing classes.6,39 Therefore, a reassessment of the complete drug history and cumulative viral genotypes and, if available, of the use of phenotypic resistance testing to determine the best possible treatment is recommended in HTE-PLWH on a failing ART.1,6,40
Long-acting LEN administration is particularly attractive for its possible use within simplification strategies in HTE-PLWH with controlled HIV viraemia.30 Multi-experienced PLWH are often treated with regimens containing a high number of drugs, resulting in a substantial pill burden and a non-negligible proportion of AEs. However, further studies are needed before this strategy can be used among HTE-PLWH in clinical practice.
Conclusions
LEN is the first-in-class CA inhibitor to be approved for the treatment of HTE-PLWH on failing ART regimens that cannot be successfully treated with other available treatments due to resistance, intolerance or safety concerns. Clinical trials have demonstrated the high efficacy and overall safety of LEN for those in this population who harbour a multidrug-resistant virus.
Given its characteristics, LEN may play an important role in the treatment of HTE-PLWH in the coming years. As with other drugs of new classes (e.g. fostemsavir and ibalizumab), However, LEN must be used appropriately to maintain its long-term efficacy in this population.